Final Project
Report
Identification of Functional Site in Proteins
Anne Christian
Rice University
Summer, 2002
The function of a protein is typically determined by the shape and chemical composition of its active site. The active site is the interface of the protein to other molecules. My project involved finding known functional sites in proteins of unknown function. The identification of a functional site determines the role of the particular protein. In this project, the templates of the functional sites were discovered by a technique called Evolutionary Trace, and the matching was done using a technique known as geometric hashing. Below I describe these in detail, after giving a small introduction on proteins. Specific details of my work follow.
Proteins- A Review
Amino acids are the mono-numeric units of a protein. [1] All amino acids have a carboxyl and an amino group connected by a central carbon atom. [2] They differ in their side chains, creating a total of 20 different amino acids commonly found in proteins. These 20 amino acids combine through peptide bonds, where the carboxyl group of one amino acid and the amino group of a second amino acid connect, releasing a water molecule in the process. [3] This process is repeated to form the chain that makes up a protein. As the amino acids combine, they begin to fold according to the DNA of the protein they are forming. Theoretically, the 20 amino acids can combine to form more proteins than there are atoms in the known universe. [4]
Proteins can perform several functions including connecting with ligands, other proteins, and various other molecules. Hemoglobin, for example, carries oxygen in the blood by binding to oxygen molecules. [5] The sequence of amino acids and the final three-dimensional shape of the protein determine the function of a protein. Proteins lock to ligands like flexible puzzle pieces. If the shape of the protein or the ligand changes too significantly at the interface, the two pieces of the puzzle will no longer fit. Although the connection site is flexible, the structure cannot contain any significant differences in shape. However, two puzzle pieces of different material will often connect to the same third piece as long as the two original pieces share a similar shape at the interface where the connection is made. The same is true of proteins. Two proteins of different amino acid sequences could connect to the same ligand equally well as long as the shape of the active site is close enough. The active site of a protein is the area directly around the docking point of the ligand. The active sequence is able to squeeze to some extent, but this squeezing is small in magnitude compared to structural changes. These small changes can be accounted for by using thresholds; however, any large, structural changes that affect the active site will cause the proteinís function to change. Variations of shape on areas outside the active site can have little or no effect on the proteinís ability to perform its task. To compare the functionality of two proteins, we will compare the chemical and structural composition of the active site.
GeoHash
GeoHash is a program written by Brian Chen, a Ph.D. student at Rice University, to determine the function of a target protein by identifying in the target protein the active site of a source protein of known function. Similar active sites imply similar functionality for the proteins, i.e., the ability to bind to similar ligands. Before the two proteins can be compared, an active site must be identified for the first protein. GeoHash employs a theory called Evolutionary Trace to create a motif of the most significant residues of the source protein. This motif is then compared to the entire target protein using Geometric Hashing, a technique developed for matching objects against a database of other objects. [6] Once the two proteins have been compared, GeoHash returns the best 20 matches.
Evolutionary
Trace
Evolutionary Trace (ET) is a technique developed by Olivier Lichtarge, M.D., Ph.D., an Assistant Professor in the Department of Molecular and Human Genetics at the Baylor College of Medicine. Evolutionary Trace is intended to determine the active sites in proteins by comparing their structure to proteins of similar ancestry. GeoHash is primarily concerned with the active site residues related to the binding site of proteins, since their shape is the most significant factor in determining a proteinís function.
In determining a proteinís active site, the evolutionary trace analyzer compares a set of proteins with similar functions but in different species. The analyzer makes two assumptions about these protein groups. First, proteins of similar ancestry must have a backbone deviation of no more than two angstroms. [7] It also assumes that any mutations made to the protein are under pressure from an evolutionary standpoint. [8] If, for example, a mutation is made to a functionally significant amino acid, the protein may no longer connect to its intended ligand. Since the proteinís shape has changed, it will be unable to propagate the mutation while maintaining the same function. The protein may, however, be able to survive by performing a different function. At the same time, if a mutation is made to a less significant amino acid, the protein will be able to continue performing the same function. As a result, proteins sharing similar backgrounds and functions should have similar active sites. [9] By observing mutation rates and functions in proteins of the same family, Evolutionary Trace can determine which atoms are functionally important.
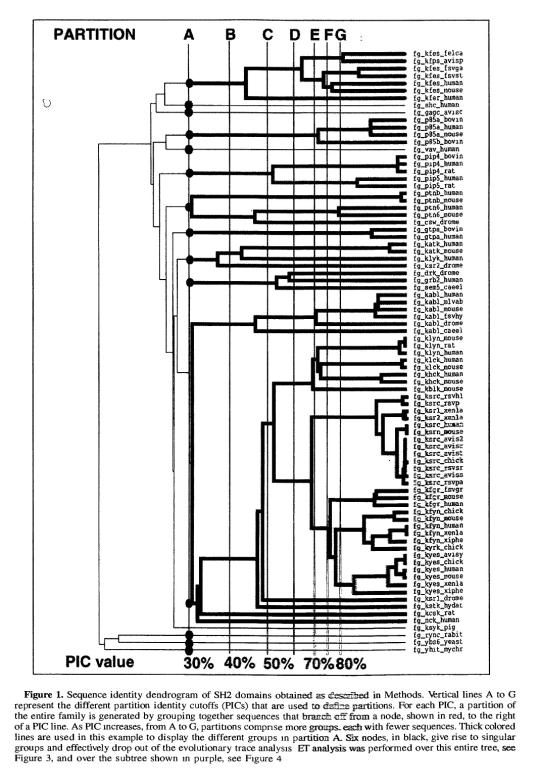
A dendrogram compares proteins of the same function, which are found in different species and expected to have become different through evolution. The proteins are sorted according to percentage sequence identity cutoffs (PIC's). At the left of the graph (Figure 1), at low PIC, the proteins are grouped into large clusters with very little information or distinction on the structure of the protein. At the same time, an extremely high PIC represents increasingly specific structure. [10] Evolutionary Trace uses the dendrogram to find related proteins. ET then finds the amino acids that are shared between these proteins. These amino acids have been shown to cluster around the active site.
Evolutionary Trace determines the active site of a protein by comparing the amino acid sequences of two proteins. First, a string of letters representing the amino acids or residues of that proteinís sequence is assigned to each protein. Then, each of these strings is pulled apart and pushed together until matches can be formed with the strings of other, related proteins. [11] This process is called alignment. Consider now the aligned proteins. If the same residue is found at the same position in all proteins that residue is given a rank one, and its symbol is placed in the appropriate location of the consensus sequence. This residue is assumed to be important to the function of the protein because of its clear lack of mutations. If a residue is found in most of the proteins, it is given a slightly larger rank, and if a residue is found in almost none of the proteins, it is given an extremely large rank. When the consensus sequence is constructed, if a residue is found in every protein at that position, but the atoms differ, then the position is represented by a neutral mark. If any of the proteins do not contain a residue in that position, then a blank is inserted into the consensus sequence. [12]
To support the theory of Evolutionary Trace, studies were then done to determine if the residues identified at high rank were in fact near the ligandís connection point. It was found that the residues were, indeed, near the point, supporting the theory of evolutionary trace. [13] Other studies were also conducted which determined the functionally relevant residues by hand, then compared them to those found through ET on the same proteins, and once again the findings of the ET were verified. We use the identified residues and their three-dimensional positions (obtained when the residues are mapped to the known 3D structure of one of the proteins involved in their determination) as a motif, a pattern, of the active site. We proceed to describe how to identify this motif to 3D structures of other proteins.
Geometric Hashing
Geometric hashing allows a computer to more easily recognize 2 and 3D objects by comparing these objects to a previously defined database of known objects. [14] To illustrate Geometric Hashing, take the example of a square. Previously, very simplistic algorithms would calculate the pixels on a known square and then traverse a screen pixel by pixel comparing the known points to the screen until another square was found. In geometric hashing, two points on the square are chosen, and a graph is drawn from those two points. The graph is then scaled so that the distance between the two points is equal to 1. An x-axis is used to connect the two points and a y-axis is drawn at the midpoint between the two points. At this time, a third point can be found in the square and plotted under the parameters of the newly formed graph. The points are then stored in a hash table with three parameters, the model number, and the x and y coordinates of the third point. The same computation is done for all of the pictureís pairs of points. All of this can be done separately, and should not affect the running time of the algorithm.
Once the database has been formed, any given object can be compared to the stored values, making match recognition easy. By plotting points on an object just as the plots were pointed on objects to form the database, a hash entry can be determined. The new entry can then be compared to the database. Each figure that matches receives a point. The process is repeated for every pair of points in the objects. Finally, the number of points each known shape received can be tallied. This algorithm does not necessarily pinpoint exactly which shape it has recognized, but it does return a list of similar shapes.
GeoHash applies geometric hashing to proteins in much the same way. The residues in the source proteinís motif are hashed into a database as points in space. The target protein is then compared to these points to determine any similar shapes. All matches meeting a previously defined criteria can then be returned to the user. My job for the summer was to work with the GeoHash program, understand its operation, and test the thresholds used to assure that the best results are returned.
Getting Started
Before I could begin my project for the summer, I did some extensive background reading. A complete list of the papers I read is included in the Works Cited. The papers were chosen based on three main topics. The first group was intended to further my understanding of protein composition and included excerpts from textbooks such as Introduction to Protein Structures. I studied proteins using Rasmol, [15] which is protein visualization software. The second group of papers focused on Evolutionary Trace, and the third group related to Geometric Hashing. Once these initial goals for the summer were achieved, I was assigned two major goals.
Goal 1: To tune methods to work faster and more reliably, and offer suggestions on improvements and changes to make to GeoHash
Goal 2: Automate the code to prepare it for a large data set experiments.
In order to accomplish the these goals, I used proteins and active site motifs from the following sets:
1. The PDB
The PDB (Protein Data Bank) is a website data bank of protein structures. Over the past thirty years, the number of protein structures in the website has increased to over 6,000. Unfortunately, such a large data set makes it difficult to thoroughly test GeoHash.
2. The "Gold" Standard
To test GeoHash, Brian Chen, a Ph.D. student at Rice University and David Kristensen, a Ph.D. student at Baylor College of Medicine have compiled a gold standard set of tests. Three groups of proteins have been compiled to test GeoHash in depth. They are designed to compare GeoHashís results to known results, to verify that GeoHash can detect misses as well as matches, and to compare GeoHashís results to previous work.
3. The L Set
The L set is a group of well-documented proteins compiled in part because of their similar function. Each protein in the set connects to a ligand. A residue cannot be included in the source motif unless it falls within four angstroms of the binding site. This helps assure that the residues are significant in the proteinís ability to connect to a ligand. Because the L set source and target proteins had similar functions, GeoHash should hypothetically find matches.
4.
The "Negatives Set"
The L set was also used to test GeoHashís ability to identify non-matching proteins. One source from each function was compared to one target from every other function. Because the sources and targets have different functions, they should also have different shapes and sequences. Ideally, under such circumstance, GeoHash should return few if any matches.
5. The TESS Set
Janet Thornton and Andrew C. Wallace compiled the TESS [16] set of proteins while working on a similar project at the University College in London. This set was chosen specifically to compare GeoHashís results to their previous work.
The Tests
Test 1:
Calculating True Positives, etc.
This test is written in Perl. After GeoHash is run, .STAT and .GAT files are output. The .STAT files contains a list of the top twenty matches in no particular order. Each match consists of a target sequence and either a ë+í or a ë|í beneath every letter in the sequence. '|'s below the sequence indicate that GeoHash found a match at this residue. The .STAT files also contain a similar structure for the source protein, where '|' indicates a residue in the motif. The sequences are then compared to an alignment file. The alignment file contains the source and target sequences, which have been spaced to make the matches of the two line up. Unfortunately, the alignment file and the .STAT file are from different sources, which means that while they are technically the same proteins, there are some variations that had to be considered. Consequently, the .STAT file sequences are also compared to the PDB files.
Once the matches and source are aligned, true positives, false positives, true negatives and false negatives are calculated followed by specificity and sensitivity. Specificity, the ability to find true positives, is calculated with the following formula:
Specificity = TN / (TN + FP)
Sensitivity, the ability to reject false positive matches, is found using the following formula:
Sensitivity = TP / (TP + FN)
Where TN = True Negative, TP = True Positive, FP = False Positive, and FN = False Negative
The .STAT files also contain information on the running time and root mean square distance (RMSD). Eventually, all of this information is output in an Excel readable, tabbed file. Later, the entire process was automated so that GeoHash can be called with different thresholds on multiple proteins, then the .STAT files can be analyzed, and an Excel readable file made.
This automation enabled the testing of two different thresholds. The first threshold, called the query threshold, is a filtering threshold. Geometric hashing compares groups of points in space. The query threshold provides an upper bound of how far apart points can be in a group. The second threshold, called the match threshold, provides an upper bound on how similar the alignment of two groups of points must be to be considered a match.
The query threshold was tested from 0.5 angstroms to 5 angstroms at intervals of .25 angstroms. The match threshold was tested from 1.5 angstroms to 5 angstroms at intervals of .5 angstroms on the L set of proteins. These results were graphed in conjunction with run time and geometric match (the total number of matches that GeoHash found).
After the tests were run on the L Set, it was clear that specificity and sensitivity reached a peak at a match threshold of 4 and that running time increased as the query threshold increased. We also discovered that RMSD (root mean square distance) stayed below one, which is a very good result.
Test 2- Negative Tests
The L set was re-run comparing each source motif to all targets of different functions. The entire process was automated using a Perl script and a make file. The results for this test are also encouraging. Few, if any, matches are found by GeoHash, indicating that GeoHash can also detect proteins of different function.
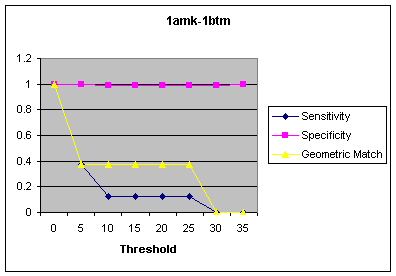
Test 3-Surface Sensitivity
Figure 2: Surface Sensitivity for source protein:1amk and target protein:1btm
One area for optimization would be to eliminate any residues not on the surface of the target. This would drastically reduce the number of comparisons run by GeoHash. To determine just how much of the residue had to be on the surface, a test was created which ran five source-target pairs at several different surface thresholds then output the sensitivities and specificities of the best matches. Unfortunately, there was a very distinct drop off in sensitivity and specificity as soon as the threshold began to exclude residues, as seen in Figure 2. The results of this test indicate that surface sensitivity would not be a good optimization because too much accuracy is lost too quickly. They also indicate that some of the atoms in the sourceís motif are buried. One idea for future work would be to re-run these tests, comparing the percentage of surface accessibility of the motif to the surface threshold.
Test 4-TESS Set
The last test to be automated was the TESS set. The TESS has a motif made up of atoms instead of amino acid residues. Before it could be run, GeoHash has to be optimized to compare atoms as well as residues. The second file output by GeoHash is a .GAT file. The .GAT file outputs a list of matches found by GeoHash. Each match contains the index numbers of all source-target pairs that GeoHash believed to be matches. The .GAT file also contains a list of all the atoms and index numbers in the source motif and target. This test, also written in Perl, uses each matchís source and target indexes to find the corresponding atoms. It then compares those atoms to see if it does in fact find a match. The best matchís (most true positive) results are then returned. This test was run at a query threshold of 2.5 angstroms and a match threshold of 2.5 angstroms. The results of this test are then output to an Excel readable file. GeoHashís results currently fall slightly short of the work done by Wallace and Thornton. One idea for future work would be to see why these results do not match and what can be changed in GeoHash to improve upon its results.
Conclusion
The tests
run helped to confirm that GeoHash runs well, without any anomalies. It also
allowed us to improve GeoHashís run time and accuracy. We were able to determine
that decreasing the query threshold could also decrease runtime without hurting
the accuracy of the results. We were also able to find an optimization for
the match threshold. This allows us to find matches for proteins that are
similar but not exactly the same, increasing GeoHashís flexibility. Because
proteins and ligands do not line up exactly, a good match threshold allows
us to find an area in which protein matches are accurate but still flexible. Although the surface sensitivity test prevented us from optimizing GeoHash,
it did provide valuable information about the type of atoms being found in
the motif. The TESS set indicated the status of GeoHash compared to previous
work and opportunities for improvement. We were also able to determine that
GeoHash could detect negative as well as positive matches. Some ideas for
further improvement include a hierarchical hashing, so that the most like
amino acids are the first to be compared. Eventually, we would like to have
code flexible enough to use on the entire PDB. Given the on-going automation
of the Evolutionary Trace and our automation efforts, we envision that our
work will be used in the future to automatically scan the PDB and isolate
those proteins that exhibit a specific motif as identified by Evolutionary
Trace. Such a capability will permit a large-scale classification of proteins.
Figure 1:[17]
Works Cited
Aloy, Patrick, Enrique Querol, Francesc X. Aviles, and Michael J. E. Sternberg.
"Automated Structure-based Prediction of Functional Sites in Proteins:
Applications to Assessing the Validity of Inheriting Protein Function from
Homology in Genome Annotation and
to Protein Docking." Journal of Molecular
Biology 2001: 311 + 395-408.
Bachar, Orly, and Daniel Fischer, and Ruth Nussinov, and Main Wolfson. "A computer
vision based technique for 3-D sequence-independent structural comparison of
proteins." Protein Engineering 1993: 279-288.
Brandon, Carl, and John Tooze. Introduction to Protein Structure. New York: Garland
Publishing, Inc., 1999.
Lictharge, Olivier, and Henry R. Bourne, and Fred E. Cohen. "An Evolutionary Trace
Method Defines Binding Surfaces
Common to Protein Families." Journal of
Molecular Biology 1996: 342-358.
Lichtarge, Olivier, Keith R. Yamamoto, and Fred. E. Cohen. "Identification of Functional
Surfaces of the Zing Binding
Domains of Intracellular Receptors." Journal of
Molecular Biology 1997: 274 + 325-337.
"Molecular Visualization Freeware." Site updated Sept. 2002
<http://www.umass.edu/microbio/rasmol/>.
Nussinov, Ruth, and Haim J. Wolfson. "Efficient Detection of Three-Dimensional
Structural Motifs in Biological Macromolecules by Computer Vision
Techniques." Proc. National Academy of Science, USA Dec. 1991: 10495-10499.
Sowa, Mathew E., Wei He, Kevin C. Slep, Michele A. Kercher, Olivier Lichtarge, and
Theodore G. Wensel. "Prediction and Confirmation of a Site Critical for Effector
Regulation of RGS Domain Activity." Nature Publishing Group 2001: 234-237.
Stryer, Lubert. "Portrait of an Allosteric Protein." New York: W.H. Freeman and
Company, 1995.
Voet, Donald, and Judith G. Voet. Biochemistry. New York: John Wiley & Sons, Inc.,
1995.
Wallace, Andrew C., and Jane Thornton. "The TESS Set."
<http://www.biochem.ucl.ac.uk/bsm/PROCAT/manual/manual.html>.
Wolfson, Hain J., and Isidore Rigoutsos. "Geometric Hashing:
An Overview." IEEE
Computational Science & Engineering 1997: 10-21.