This summer (June – August 2013), I traveled to Tufts University in Medford, MA as part of the Distributed Research Experience for Undergraduates (DREU) program. In broad terms, we improved protein function prediction through applications of graph theory and confidence in protein-protein interaction networks.
The results of this project are summed up in:
We also hope that this work will be part of a larger paper submitted sometime in 2014.
For a week-by-week account of this summer’s journey, see my blog!
Also, a huge thanks goes out to the DREU Co-Directors for selecting me as a Clare Boothe Luce Undergraduate Research Scholar for this summer’s work. I am so grateful for this honor!
I hope you enjoy my summary below! If you’re new to protein function prediction, I also included a little blurb about protein networks – scroll down below the summary to find it. Thanks!
Project Summary
For this project, we worked extensively with protein-protein interaction (PPI) networks to predict individual protein functions more accurately. More specifically, we set out to improve an algorithm called Diffusion State Distance (DSD), which takes advantage of graph diffusion to better capture the distances between two proteins. This has the effect of giving more weight to interactions within tight local networks rather than those that go through hubs or are only distantly connected.
Lenore beautifully described this problem using Facebook as a metaphor. Just as protein functions are predicted by looking at a protein's neighbors, Facebook friends are suggested by looking at mutual friends. Are you more likely to find a “real” mutual friend through Beyonce or your best friend from high school? Even if you actually know Beyonce personally, you’re still not very likely to locate people you actually know through her. Why? It's a numbers game. She has millions of friends/connections. Odds are your best friend has less than 1000. The same goes for proteins; proteins in the same true neighborhood are far more likely to have similar functions than proteins that happen to connect across a hub.
My part in this seeks to improve the algorithm even further by accounting for unreliability that is often inherent in biological databases. We do this by assigning a confidence value to each interaction. This confidence can be derived in lots of different ways – the number of literature citations backing an interaction, the experimental methods used to find the interaction, relationships between gene ontology, and many more. For example, if a particular interaction has been cited in 10 papers, that interaction is generally more reliable than an interaction cited in just one paper.
Our first goal was to simply (and somewhat naively) integrate this confidence information into the algorithm and see what happens. We did this by generating 100 random versions of the network, using the confidence values as the probability that each edge exists. It turned out that this method introduced too much noise, and the effect of adding the confidence values was not able to shine through. We determined that we’d have to scale this up by several orders of magnitude in order to identify the patterns that appeared, which is not tangible within the scope of this project.
Instead, we incorporated confidence into the random walk component of DSD. (See the DSD website for more about the internals of DSD.) Instead of weighting each path out of a node equally, these paths were weighted by the edges' confidence values, normalized with respect to the other paths leading out of the node. This worked MUCH better, and we were able to improve function prediction performance significantly.
What are Protein Networks?
Proteins are found in all living things, and serve a wide variety of different purposes depending on their structure. For example, the proteins hemoglobin and fibrinogen are both found in the human bloodstream. Hemoglobin transports oxygen, and fibrinogen forms blood clots. Although both function within the bloodstream, they are built for completely different purposes.
Here's where protein networks come in. In short, protein-protein interaction (PPI) networks describe which proteins work together. Because proteins that interact with one another are more likely to functionally relate to one another than proteins that don't, we can use these networks to predict protein functions. The graph below shows a subset of the PPI network for yeast.
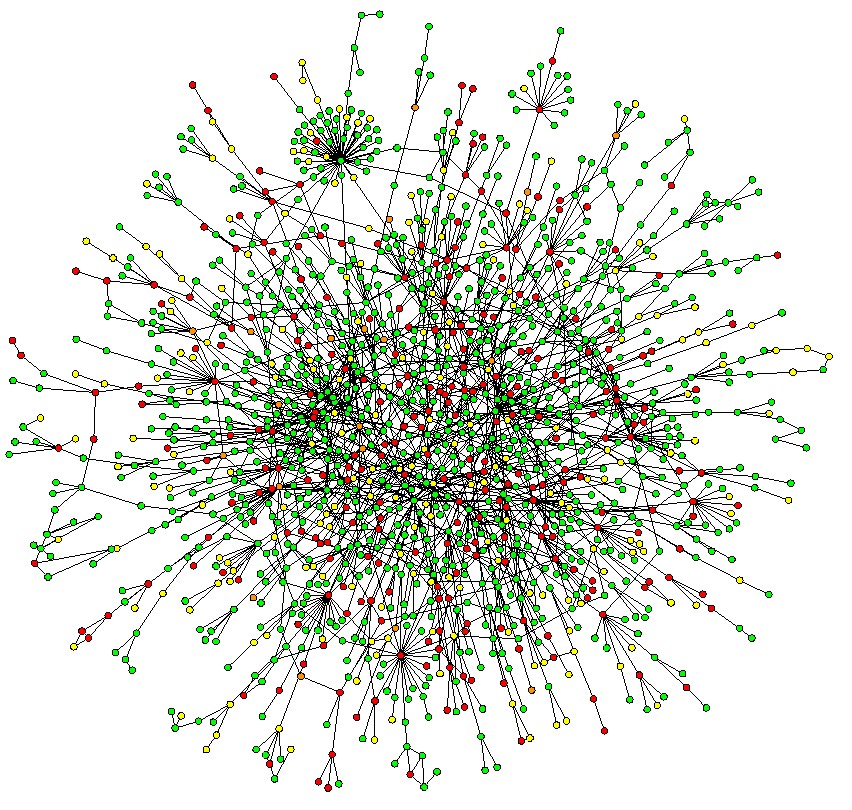
How are Protein Functions Predicted?
There are dozens of computational methods for predicting protein functions, the most accurate and popular of which calculate the shortest path between proteins. Even though such algorithms are pretty good, they do have limitations. It turns out that around half of all proteins are pretty close together at a distance of two. This usually happens because of the presence of "hubs," single nodes with huge numbers of connections. These hubs are the super-tight clusters in the graph above.